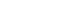The molecular mechanisms underlying retinal neural development and regeneration are complex, involving DNA variation, epigenetic modifications, RNA transcription, and splicing. Integrated multi-omics analysis is considered an effective approach to decipher the molecular mechanisms of neural stem cell lineage differentiation and regeneration. As multiple neural lineages are intermingled during development and regeneration, accurately distinguishing different lineages and systematically delineating the dynamic changes in their multi-dimensional molecular features is key to precisely deciphering these molecular mechanisms. In recent years, the development of single-cell multi-omics sequencing technologies has provided powerful methods for analyzing the complex molecular mechanisms of neuronal development and regeneration. However, RNA alternative splicing, a crucial aspect of gene function regulation, still lacks effective methods for integrated single-cell multi-omics correlation analysis.
On September 13, 2024, Professor Hu Youjin's team from Zhongshan Ophthalmic Center, Sun Yat-sen University, published a research paper online in Nature Communications titled “Simultaneous profiling of RNA isoforms and chromatin accessibility of single cells of human retinal organoids”. The paper reports a novel single-cell multi-omics sequencing method named scRICA-seq (single-cell RNA isoform and chromatin accessibility sequencing). This technology can simultaneously detect chromatin accessibility, RNA expression, and full-length RNA isoform structures at the single-cell level, enabling integrated analysis of epigenetics, transcription, and RNA splicing within individual cells. It provides technical support for uncovering new mechanisms in retinal neuron development and regeneration.
Professor Hu's team has long focused on innovating and developing single-cell sequencing technologies. As early as 2020, the team published the scRCAT-seq technology for RNA isoform detection in Nature Communications. This time, the team further improved the original method, developing the scRCAT-seq2 full-length transcriptome sequencing technology. By integrating it with the 10x Genomics Chromium Next GEM Single Cell Multiome ATAC + Gene Expression platform, they achieved a multi-omics method capable of simultaneous detection of chromatin accessibility and RNA isoforms. The core technology of scRCAT-seq2 involves processing cDNA using a single-molecule circularization technique and labeling sequencing reads covering the Transcription Start Site (TSS), Transcription End Site (TES), and exons with the same barcode, allowing for the assembly and construction of full-length cDNA sequences. (Figure 1)
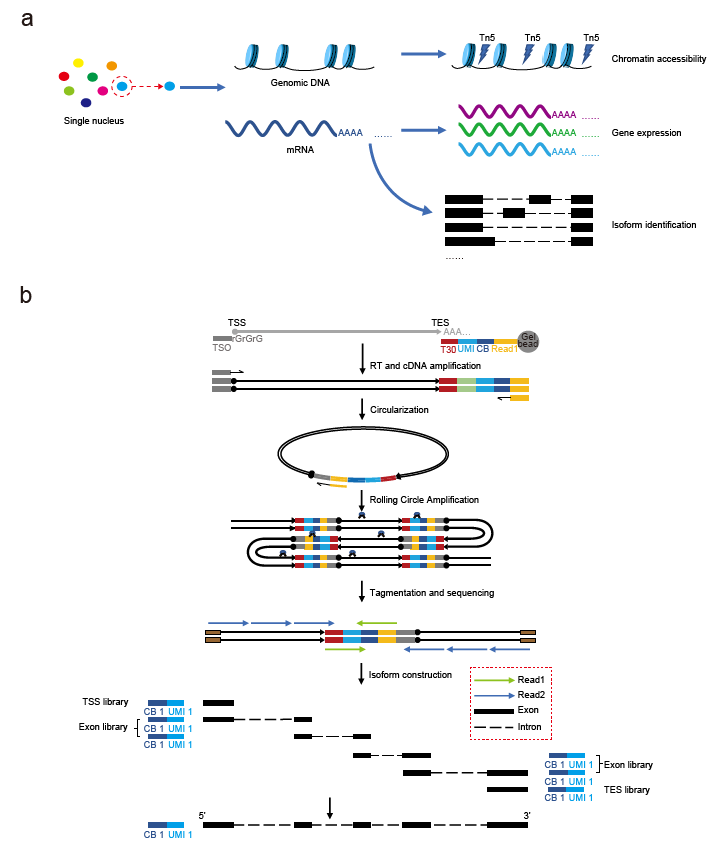
Figure 1: Schematic diagram of scRICA-seq and scRCAT-seq2
The researchers compared scRCAT-seq2 with various third-generation sequencing technologies (such as SCAN-seq2, scISOr-seq, HIT-scISO-seq, etc.) under both low- and high-throughput conditions. The results showed that scRCAT-seq2 has significant advantages in terms of cost-effectiveness and detection sensitivity. (Figure 2)
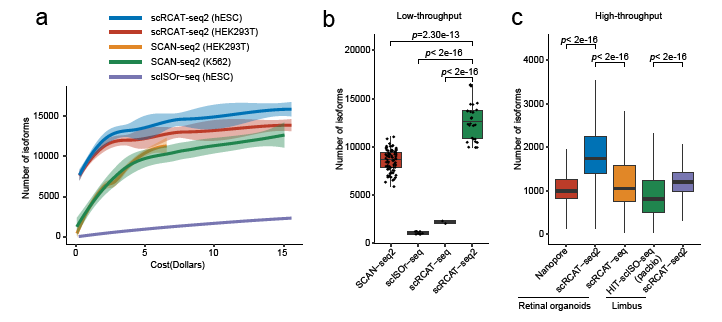
Figure 2: Comparative analysis of scRCAT-seq2 with other methods
The researchers applied scRICA-seq to the human retinal development process, constructing a single-cell multi-omics atlas of human retinal organoids. Based on single-cell isoform clustering analysis, they identified six cell clusters corresponding to six typical early retinal neurons: retinal progenitor cells (RPCs), retinal ganglion cells (RGCs), amacrine cells/horizontal cells (AC/HCs), photoreceptor precursors (PR precursors), neurogenic RPCs, and cone photoreceptors (cones). These results were highly consistent with data obtained using scRNA-seq technology. (Figure 3)
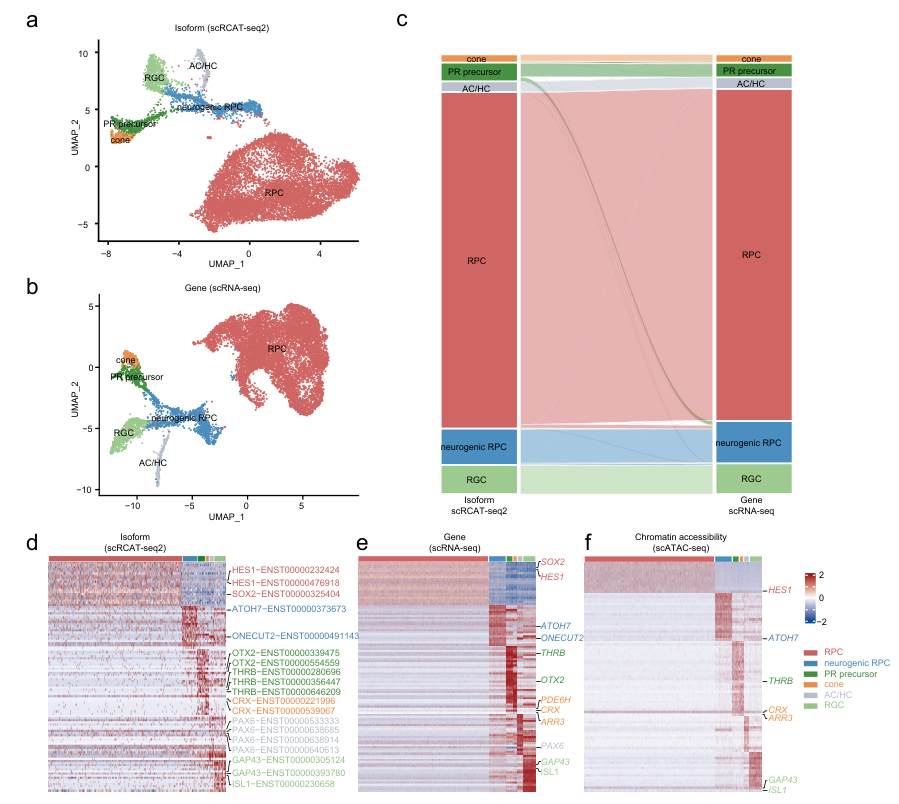
Figure 3: Multi-omics single-cell atlas of human retinal organoids based on scRICA-seq
Furthermore, the authors investigated the dynamic selection patterns of RNA isoforms during the differentiation of RPCs into cones. They identified 2292 differentially expressed isoforms between cones and RPCs, with 1143 isoforms highly expressed in cones and 1149 highly expressed in RPCs. Through transcription factor (TF) motif analysis, the researchers predicted 589 TFs that might bind to these dynamically regulated splicing sites. The predicted TFs included 11 previously reported to be involved in regulating RNA splicing, such as NEUROD1, SP1, and KLF4, as well as known cell fate determinants like NEUROD1, ASCL1, and THRB. This suggests that dynamic changes in chromatin accessibility at splicing sites and RNA splicing may cooperatively regulate cell fate decisions. (Figure 4)
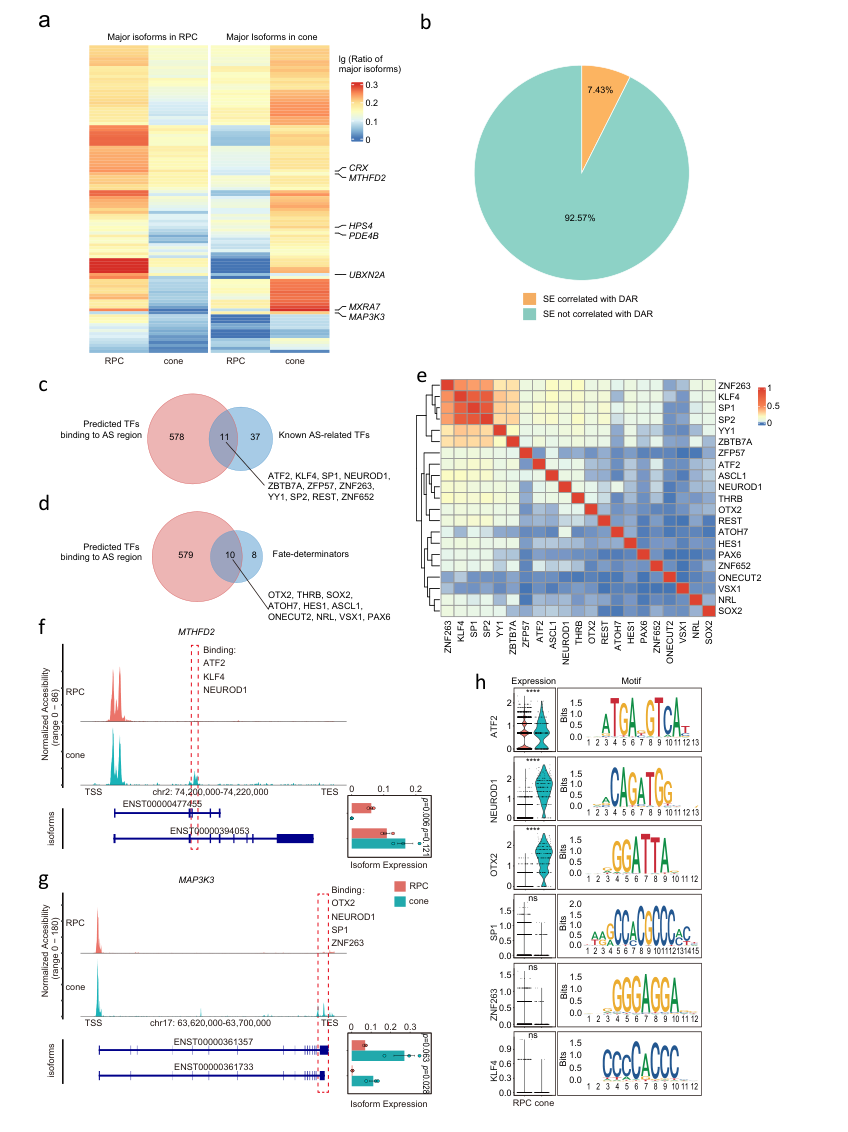
Figure 4: Correlation between RNA isoform selection, chromatin accessibility, and TFs during RPC development
In summary, scRICA-seq enables integrated analysis of epigenetics, RNA transcription, and splicing at the single-cell level. The study found that chromatin accessibility and fate-determining transcription factors cooperatively regulate downstream gene expression and transcriptional splicing, providing new insights into the mechanisms governing retinal neuron development and fate determination.
About the Research Team
This study was completed by Professor Hu Youjin's research group at Zhongshan Ophthalmic Center, Sun Yat-sen University. Zhang Shuyao (Research Assistant), Xiao Yuhua (PhD student), and Mo Xinzhi (Research Assistant) from Zhongshan Ophthalmic Center are co-first authors. Professor Hu Youjin is the sole corresponding author. Zhongshan Ophthalmic Center of Sun Yat-sen University / State Key Laboratory of Ophthalmology is the first and corresponding affiliation unit.
Link to the original article: